Cistična fibroza
Sopomenke v širšem pomenu
Cistična fibroza, pljuča
Angleščina: mucoviscidosis, cistična fibroza
Opredelitev cistične fibroze
Cistična fibroza je dedna bolezen. Dediščina se medicinsko imenuje avtosomno recesivna. Cistična fibroza (cistična fibroza) se ne podeduje na spolnih kromosomih X in Y, temveč na avtosomskem kromosomu 7.
Preberite naš splošni članek o presnovnih motnjah: Presnovne motnje - kaj to pomeni?
Mutacija je na tako imenovanem CFTR genu. Recesivno je pomenilo, da morata biti prisotni dve okvarjeni kopiji gena, da bolezen izbruhne. Če ima oseba zdravo in mutirano mesto gena na ustreznem kromosomu 7, se bolezen ne pojavi.
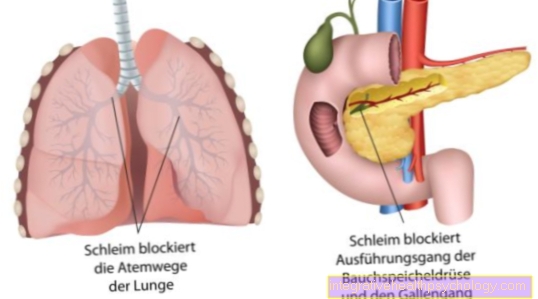
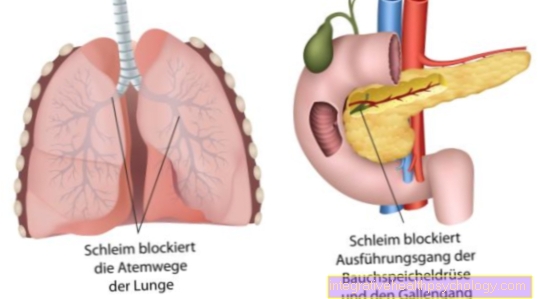
Rezultat je patološki genski izdelek. Tako kodirano Kloridni kanali so zlomljeni. Okvarjeni kloridni kanali vodijo do nastanka debele sluzi v vseh zunanjih žlezah.
Te zunanje žleze, tj. Žleze, ki izločajo izločanje navzven, vključujejo:
- trebušna slinavka
- tanko črevo
- sistem dihalnih poti s pljuči in bronhialnim sistemom
- žolčnih poti in
- tudi Žleze znojnice
Povzetek
Cistična fibroza je Dedne bolezni. Podeduje se tako, da je neodvisno od spola in samo z dva okvarjena gena nastopi. Je najpogostejša avtosomno recesivna dednost.
Posledice so težke tvorbe sluzi vseh zunanjih žlez, na primer žlez pljuč, trebušne slinavke in tudi žlez znojnic. Temeljijo na tem moten transport klorida med notranjostjo in zunaj celice (beri dalje: Klorid v krvi). Mutirani gen je vklopljen Kromosom 7 in povzroča najrazličnejše vpletenost organov z ustreznimi učinki na dihanje, prebavo in razmnoževanje.
Na žalost lahko terapija le ublaži simptome, ne pa tudi zdravljenja. The Pričakovana življenjska doba pri bolnikih s cistično fibrozo relativno nizka.
Ker gre za recesivno dedno bolezen, obstajajo ljudje, ki nosijo spremenjen gen, vendar ne trpijo zaradi same bolezni. Takšne osebe so poklicane Nosilec funkcije ali Dirigenti, to je prevoznikov. Ti ljudje nimajo cistične fibroze, ker je druga kopija gena nedotaknjena in bolna ni dovolj močna, da bi lahko prevladala.
Vendar lahko to pomanjkljivo kopijo gena posreduje svojim potomcem. Če bi spremenjeni gen že zadostoval za nastanek bolezni, bi šlo za tako imenovano prevladujoče dedovanje. Takšno dedovanje lahko najdemo na primer v Chorea hunton. Več o tej bolezni lahko izveste v naši temi Chorea hunton.
Približno 1:2500 se skriva Stopnja bolezni pri novorojenčkih v Nemčiji. Prevoznik gre za vsakogar 25. pri nemškem prebivalstvu.
glavni vzrok

Cistična fibroza povzroča mutacijo gena na kromosomu 7. Ta kromosom je avtosomski kromosom in ne spolni kromosom.
Vsakdo ima 44 avtosomskih kromosomov (dve identični različici vsakega) in dva spolna kromosoma. Ta mutacija na kromosomu 7 vodi do nastanka okvarjenih kloridnih kanalov. Ponovna absorpcija (ponovna absorpcija) klorida iz žleznih izločkov ni mogoča, ker receptor, priklopna točka za klorid, ni vgrajen v žlezne kanale.
Namesto tega je zaradi nepravilnega videza in strukture odložena za rudarjenje. Naravna izmenjava klorida skozi določene kloridne kanale je motena. Ti tako imenovani kanali so sestavljeni iz beljakovin. Na naši DNK je kodirana najrazličnejša beljakovina. Zaradi genetske okvare kloridnih kanalov prihaja do dehidrirane in žilave tvorbe sluzi iz vseh žlez, ki izločajo izločanje navzven. Sluz nato delno blokira kanale ali dihalne poti v pljučih.
Preberite tudi o tem Mutacija kromosomov
Diagnoza cistične fibroze

Značilni simptomi, ki se začnejo v povojih, so prelomni pri diagnozi cistične fibroze.
Ta sum okrepi pozitivna družinska anamneza (bolezen očeta / matere ali bližnjih sorodnikov). Pozitivna družinska anamneza pomeni, da obstajajo ali so že bili primeri cistične fibroze znotraj družine - po materinski ali očetovi strani.
Pomanjkanje encimov trebušne slinavke je mogoče zaznati tudi v blatu. Morebitne blokade dihalnih poti lahko odkrijemo z rentgenskim slikanjem prsnega koša.
Test znoja, ki meri vsebnost klorida v znoju, pomaga tudi pri diagnozi cistične fibroze. Če je določena vrednost presežena in se uporabljajo tudi drugi simptomi, je diagnoza razmeroma fiksna. Pogosto starši sami opazijo povečano vsebnost soli v znoju dojenčka.
Nerojeni otrok se lahko tudi testira na to dedno bolezen. Uporaba punkcije amnijske tekočine (Amniocenteza) celice ploda se odstranijo in pregledajo za mutirani gen.
Preberite več o temi: Rentgenski pregled otroka
Terapija cistične fibroze
Vsi, ki jih je prizadela cistična fibroza, bodo v enem prejeli nasvet Cistična fibroza - ambulantni oddelek ali nasvet od Človeški genetik (Specialist za dedne bolezni) priporočljivo. Te lahko pomagajo povečati kakovost življenja ali, če želite imeti otroke, izračunajte verjetnost obolelega otroka. Pod pogojem, da so starši rodovitni in rodovitni.
V nasprotnem primeru je zdravljenje simptomatsko, saj vzroka, okvarjenega gena, ni mogoče odpraviti.
Nalezljiva bolezen
Cistična fibroza (cistična fibroza) je še danes neozdravljiva bolezen.
V primeru cistične fibroze je pomemben ustrezen vnos namizne soli (Natrijev klorid, NaCl). Seveda je namenjena mukoliza. Mukoliza je raztapljanje sluzi, zlasti v pljučih, da olajšamo dihanje.
Zdravila in inhalacije lahko ublažijo simptome. Če se funkcija pljuč opazno poslabša, lahko dajemo kisik.
Z intenzivno fizioterapijo (fizioterapija), na primer masaže s tapkanjem in dihalnimi vajami, se zdravijo tudi spremembe pljuč, ki jih povzroča cistična fibroza.
Pogosto se bolezen konča z zahtevano presaditvijo pljuč. Vendar so čakalne liste dolge.
Del terapije je tudi peroralno dajanje encimov trebušne slinavke in vitaminov, topnih v maščobi. Nalogo trebušne slinavke je zato treba podpreti ali bolje zamenjati. V maščobi topni vitamini so A, D, E in K. Dati jih je treba neposredno v kri, saj jih zaradi pomanjkanja prebavnih encimov ne moremo absorbirati iz hrane.
Prehrana mora biti tudi visoko kalorična, saj jih lahko le del pridobimo iz hrane.
Da bi se izognili dodatnim dejavnikom tveganja za zaplete, kot sta gripa ali pljučnica, je treba otroka cepiti. Priporočajo se naslednja cepljenja:
- ošpice
- Pnevmokoki
- gripe
Preberite več o temi: Superinfekcija
Seveda je za te ukrepe potrebno posvetovanje z zdravnikom, s katerim se je treba pogovoriti o tveganjih.
Danes je veliko upanja na zdravljenje cistične fibroze v genetskih raziskavah. Poskuša se vnesti manjkajoča genetska informacija v človeški genom. Iščemo vektorje, ki bi lahko obvladali to nalogo. Vektorji so lahko na primer bakterijska ali virusna DNK, ki zdravo frekvenco uspejo vključiti v naš genetski sestav.
Trenutno se preizkuša terapevtski pristop pri nerojenih bolnikih. Pri miših so mišji zarodki že uspeli pretihotapiti zdravi gen, ki je vseboval pravilno gensko zaporedje, in sicer z amniocentezo (inokulacijo amniotske tekočine). Zdravi gen CFTR je bil torej proizveden v teh miši. Amniocenteza je punkcija in odstranjevanje otrokovih celic iz amnijske tekočine, to pa poteka skozi materino steno trebuha.
V Nemčiji pa je ta oblika intrauterine (= v maternici = v maternici) prepovedana "terapija".
profilaksa
A preventivni ukrep v tem smislu ne obstaja, ker gre za dedno bolezen.
Lahko pa obiščemo človeško genetsko svetovanje (običajno ga najdemo v univerzitetnih bolnišnicah). Tu se izračuna, kako visoko bi bilo tveganje za prenos bolezni na otroke.
Ta nasvet je vedno koristen, če obstaja družinska anamneza cistične fibroze.
Tudi eno Prenatalna diagnostika vredno si je prizadevati. Tu pred rojstvom (t.j. prenatalno) a Pregled amnijske tekočine (Amniocenteza) izvede. Fetalne celice (otrokove celice) se odvzamejo iz amnijske tekočine in DNK se pregleda za mutiran gen.
Prognoza cistične fibroze
Žal je povprečna življenjska doba pri bolnikih s cistično fibrozo le 32-37 let. Danes je pričakovana življenjska doba novorojenčkov, rojenih s tem stanjem, približno 45–50 let.
Prognoza je močno odvisna od terapije in od tega, ali se je drži.
Pacient sam in njegova motivacija imata zato pomembno vlogo.